1. Электролитический метод
Сущность метода. Метод основан на выделении меди электролизом на платиновом сетчатом катоде в азотно-сернокислом растворе при силе тока 1-1,5 а и напряжении 2-3 в. Определение заканчивают весовым методом по количеству выделившейся меди.
Электролитическое выделение меди в присутствии больших количеств трехвалентного железа невозможно вследствие окисления металлической меди трехвалентным железом. При анализе материалов, содержащих железо, мышьяк и другие мешающие компоненты, электролитический метод определения меди применим только после предварительного отделения ее тиосульфатом натрия или другими реагентами.
Выполнение определения. Навеску 1 г образца помещают в стакан емкостью 300 см3, прибавляют 15 см3 свежеприготовленной серной кислоты (1:1), 5 см3, азотной кислоты (плотность 1400 кг/м3) и растворяют сначала на холоду, затем осторожно нагревают до полного растворения. Обмывают стенки стакана водой и кипятят до удаления окислов азота. Разбавляют раствор горячей водой до объема 150 см3 и нагревают до кипения. Раствор кипятят 1 мин и ставят на электролиз. Электролиз проводят с применением сетчатых платиновых электродов при силе тока 1,5 а в течение 3-4 ч. Для проверки полноты выделения меди приливают 20 см3 воды и продолжают электролиз еще в течение 15 мин. Отсутствие меди на свежепогруженных частях катода указывает на окончание электролиза.
Не прерывая тока, электроды промывают последовательно в двух стаканах с водой, затем выключают ток, катод промывают спиртом, высушивают его при температуре 105-110°С в течение 5-7 мин, охлаждают в эксикаторе и взвешивают. Процентное содержание меди вычисляют по формуле:
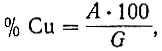
где А - вес осадка металлической меди, г;
G - навеска образца, г.
В электролите определяют медь фотоколориметрическим пиридино-родановым методом. Для этого поступают следующим образом.
Электролит выпаривают до паров серной кислоты, охлаждают, прибавляют 10 см3 азотной кислоты (плотность 1400 кг/м3) и снова выпаривают до паров серной кислоты. Соли растворяют в 20 см3 катионированной воды и фильтруют в мерную колбу емкостью 50 см3, разбавляют до метки катионированной водой и перемешивают. Берут аликвотную часть 10 см3, помещают в градуированную пробирку с притертой пробкой, прибавляют 1 см3 раствора лимонной кислоты (500 г/л), 2 капли фенолфталеина и нейтрализуют, прибавляя по каплям раствор едкого натра (250 г/л) до появления розовой окраски. Затем подкисляют 1-н. раствором серной кислоты до исчезновения окраски. Прибавляют 6 см3 раствора роданистого аммония (100 г/л), 1 см3 пиридина и 10 см3 хлороформа. Хорошо встряхивают и дают отстояться. Хлороформенный слой сравнивают со стандартным раствором меди методом уравнивания окраски. Для этого в одну из пробирок прибавляют все реактивы и затем стандартный раствор меди до одинаковой окраски с испытуемым раствором. Найденное количество меди приплюсовывают к основному, полученному путем электролиза.
Сущность метода. Метод основан на выделении олова дегерированием в азотнокислом растворе в виде β-метаоловянной кислоты с последующим фильтрованием и промыванием.
Реакция протекает по уравнению:
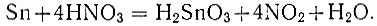
Осадок β-метаоловянной кислоты растворяют в горячем растворе едкого натра.
Уравнение реакции:
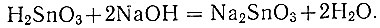
Адсорбированные β-метаоловянной кислотой небольшие количества меди осаждают сернистым натрием, растворяют и присоединяют к основному раствору.
Выполнение определения. Навеску 1 г образца помещают в стакан емкостью 300 см3, прибавляют 30 см3 азотной кислоты (плотность 1400 кг/м3) и растворяют при умеренном нагревании. По окончании растворения стенки стакана обмывают водой и раствор выпаривают до выделения солей. Соли растворяют в 2 см3 азотной кислоты (плотность 1400 кг/м3), разбавляют раствор горячей водой до объема 150-200 см3, прибавляют немного бумажной массы, кипятят 2-3 мин и выдерживают на теплой плите в течение двух часов. Выделившуюся метаоловянную кислоту фильтруют на плотный фильтр и промывают 8-10 раз горячим раствором азотной кислоты (3:97). Фильтрат сохраняют. Фильтр с осадком помещают обратно в стакан и растворяют олово в 100 см3 горячего раствора едкого натра (100 г/л). Прибавляют 100 см3 горячей воды, 15 см3 раствора сернистого натрия (50 г/л) и кипятят 2-3 мин. Дают осадку хорошо отстояться и затем фильтруют на фильтр средней плотности. Осадок промывают 6-8 раз холодным раствором сернистого натрия (10 г/л). Осадок помещают в фарфоровый тигель, озоляют и прокаливают при температуре 500-600°С. Остаток растворяют в тигле с 3-5 см3 азотной кислоты (плотность 1400 кг/м3), разбавляют 3-5 см3 воды и фильтруют. Полученный раствор присоединяют к основному.
Раствор нейтрализуют аммиаком (плотность 900 кг/м3) до выпадения основных солей меди, затем снова подкисляют азотной кислотой (плотность 1400 кг/м3) до растворения образовавшихся солей. К раствору прибавляют 4 см3 азотной кислоты (плотность 1400 кг/м3), 4 см3 серной кислоты (1:1), 10 см3 азотнокислого аммония (500 г/л), разбавляют раствор водой до объема 200 см3, нагревают до кипения и кипятят 1 мин. Выделяют медь электролизом при силе тока 1 а в течение 12-14 ч.
Электрод должен быть полностью покрыт раствором во избежание окисления меди.
По окончании электролиза электролиты выпаривают до объема 5-10 см3 и разрушают аммонийные соли. Для этого прибавляют 25 см3 азотной кислоты (плотность 1400 кг/м3) и 25 см3 соляной кислоты (плотность 1190 кг/м3), закрывают стакан стеклом и кипятят раствор до объема 10-15 см3 Операцию повторяют и затем выпаривают раствор до паров серной кислоты. Растворяют соли в воде и проверяют на присутствие меди фотоколориметрическим пиридино-родановым методом, как описано в варианте А.
Процентное содержание меди вычисляют по формуле (10).
|
ПОИСК:
|
При использовании материалов сайта активная ссылка обязательна:
http://metallurgu.ru/ 'Библиотека по металлургии'